2.4. Intermolecular Potentials#
In addition to nonbonded interactions (e.g. Lennard–Jones and Coulomb), molecular simulations must describe the internal geometry of molecules: how atoms are held together by covalent bonds and how those bonds can bend and twist. These “bonded” or “intramolecular” interactions are usually written in terms of:
bond lengths between pairs of atoms \(i\) and \(j\),
bond angles between triplets \(i,j,k\), and
dihedral (torsional) angles between quadruplets \(i,j,k,\ell\),
in Fig. 2.12 most typical intermolecular potentials for a molecule are shown.
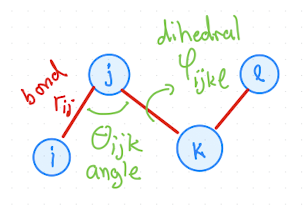
Fig. 2.12 Visualization of bonds, angles, and dihedrals in a molecule with four atoms.#
The total bonded intramolecular energy is conventionally written as:
where:
\(r_{ij} = \lVert \mathbf r_i - \mathbf r_j \rVert\) is the bond length,
\(\theta_{ijk}\) is the angle at atom \(j\) between bonds \(ij\) and \(jk\), and
\(\varphi_{ijkl}\) is the dihedral angle defined by the two planes \((i,j,k)\) and \((j,k,\ell)\).
For molecular dynamics, we do not only need energies but also forces. For any potential \(U({\mathbf r_i})\) the force on atom \(i\) is given by:
For bonded terms this is slightly more involved than for a simple pair potential, because we must use the chain rule:
we differentiate \(U\) with respect to \(r_{ij}\), \(\theta_{ijk}\), or \(\phi_{ijkl}\), and then differentiate those geometric quantities with respect to the atomic coordinates.
In actual simulations under periodic boundary conditions, distances such as \(r_{ij}\) are almost always computed with a ``minimum image’’ distance function, so that a bond crossing a periodic boundary still has the correct length.
To compute a conservative force in general: \( \vec{F}_i = -\nabla_i U \) where \(\nabla_i = \frac{\partial}{\partial r_i}\) is the gradient with respect to \(r_i\).
2.4.1. Bond Potentials#
As shown in Fig. 2.13, the bond length between particle \(i\) and \(j\) is given by
\( r_{ij} = \| \vec{r}_i - \vec{r}_j \| = \sqrt{(x_j-x_i)^2 + (y_j-y_i)^2 + (z_j-z_i)^2}\).
The bond vector is \( \vec r_{ij} = \vec{r}_i - \vec{r}_j \).
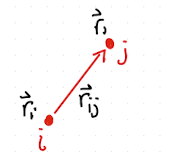
Fig. 2.13 Definition of bond distance.#
To obtain the force on a bond we need to compute
For this, we need \(\nabla_i r_{ij}\), which is
which is the unit vector pointing in the direction of the bond!
The force then is
By symmetry we should have \(\nabla_j r_{ij}=-\nabla_i r_{ji}\), meaning that
This shows that for a bond, \(F_i = -F_j\), which illustrates that action has a equal and opposite reaction.
Thus the bond force is always collinear with the bond and equal and opposite on the two atoms — a direct manifestation of Newton’s third law. In code, one can compute the force once and apply \(\mathbf F_i\) and \(-\mathbf F_i\) to save work.
Tip
Once can use the action reaction principle to save on computations by only computing either \(F_i\) OR \(F_j\) and then applying the equal action-reaction principle to save on calculations.
2.4.1.1. Harmonic#
\( U(r) = \frac{1}{2}k(r - r_0)^2 \)
Here:
\(r_0\) is the equilibrium bond length,
\(k_b\) is the force constant (spring constant) with units of energy / length\(^2\).
This fixes the bond length to fluctuate around an average of \(r_0\).
Watch for factor of 2!
Typically \( k \sim 10^3 \epsilon/l^2\) for “stiff” bond
This potential has a single minimum at \(r_{ij} = r_0\) and grows quadratically as the bond is stretched or compressed. The factor of \(1/2\) is conventional and very convenient: when we differentiate to obtain the force, the factor \(2\) from the exponent cancels the \(1/2\) in front, so \(k_b\) directly controls the strength of the force.
Example: Harmonic bond force
\( U_b = \frac{1}{2}k(r - r_0)^2 \), so
\( F_b = \frac{\partial U_b}{\delta r_{ij}} = -k (r_{ij}-r_0)\), leading to
If \(r_{ij}>r_0\), the bond is streched past \(r_0\) and particle \(i\) moves toward \(j\), and \(j\) toward \(i\). If \(r_{ij}<r_0\), the bond is compressed below the set point \(r_0\) and particle \(i\) moves away from \(j\), and \(j\) away from \(i\).
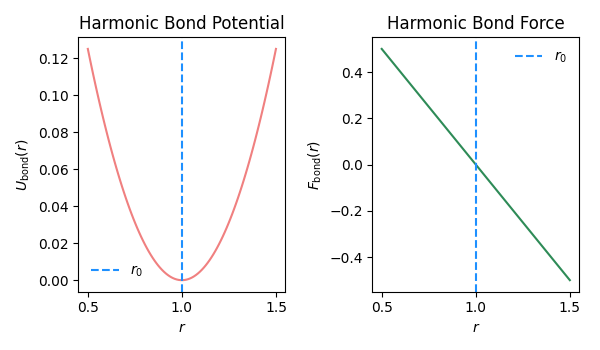
Fig. 2.14 Bond potential and force as function of bond length \(r\).#
Physical picture.
If \(r_{ij} > r_0\), the bond is stretched: \(r_{ij} - r_0 > 0\), so \(F_b < 0\) and the force pulls the atoms \emph{toward} each other to restore the equilibrium distance.
If \(r_{ij} < r_0\), the bond is compressed: \(r_{ij} - r_0 < 0\), so \(F_b > 0\) and the force pushes the atoms \emph{apart}.
Example.
In an all-atom force field, a typical stiff covalent bond might use \(k_b \sim 10^3\) (in reduced units) so that the bond length fluctuates only slightly around \(r_0\) during the simulation.
2.4.1.2. FENE (Finite Extensible Nonlinear Elastic)#
For polymers and coarse-grained models it is often desirable to prevent bonds from stretching arbitrarily far. The FENE potential does exactly this:
\( U(r) = -\frac{1}{2}k r_0^2 \ln\left(1 - \left(\frac{r}{r_0}\right)^2\right) \)
Diverges at \(r_0\), minimum at 0. This is usually used in conjuction with a repulsive, short-ranged force (WCA = cut LJ at minimum and shift up to get a purely repulsive potential).45
with parameters:
\(k_f\) — FENE spring constant,
\(R_0\) — maximum bond extension.
Key properties:
\(U_b^{\text{FENE}}\) has a minimum at \(r_{ij}=0\).
As \(r_{ij} \to R_0\), the logarithm diverges and the potential goes to \(+\infty\): the bond cannot extend beyond \(R_0\) (finite extensibility).
For moderate extensions (\(r_{ij} \ll R_0\)), the potential is roughly quadratic and behaves similarly to a harmonic spring.
In practice FENE bonds are almost always combined with a short-range repulsive nonbonded potential (e.g.\ a Weeks–Chandler–Andersen or purely repulsive Lennard–Jones) so that the overall interaction has a finite minimum at a nonzero distance. A widely used parameter set in coarse-grained polymer simulations is :
\(k_f = 30\), \(r_0 = 1.5\)
(in reduced Lennard–Jones units), which allows bonds to stretch up to about 1.5 times their typical length before the potential diverges.
Example. In the Kremer–Grest polymer model, beads connected by FENE+WCA bonds behave like flexible springs that can fluctuate but never break, making them ideal for studying polymer melts and entangled chains.17
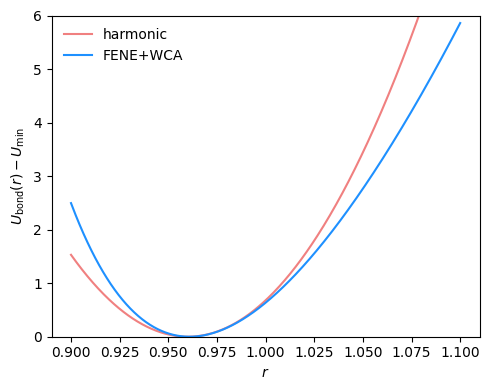
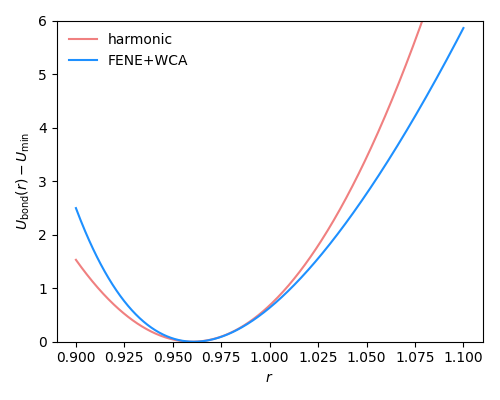
Fig. 2.15 FENE+WCA bond potential with standard parameters \(R_0=1.5\) and \(k=30\) compared to a harmonic bond potential with \(r_0=0.96\) and \(k=850\).#
2.4.2. Angle Potentials#
The angle between three particles \(i\), \(j\), and \(k\) is given by:
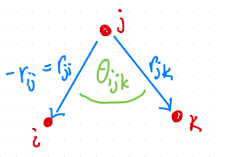
Fig. 2.16 Definition of angle.#
Note the negative sign, i.e the two bond vectors point out from the middle particle \(j\).
Note
In polymers, angle θ is sometimes defined as the outer angle. This is less common in atomistic models, but be careful!
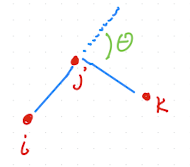
Fig. 2.17 Alternative definition of angle as the outer angle.#
These two definitions can be easily converted into each other \(\theta_{ijk} = \pi - \theta'_{ijk}\). This is just a different convention, but it matters when specifying equilibrium angles and when interpreting the potential. Computing \(\theta_{ijk}\) explicitly requires an \(\arccos\) operation, which is relatively expensive. Many force fields therefore write angle potentials directly in terms of \(\cos\theta_{ijk}\) to avoid calling \(\arccos\) inside tight inner loops.
To compute the angle potential between three particles, we need to:
Getting \(\cos^{-1}\) is computationally expensive, which would be needed above. Once can try to avoid that by using the chain rule:
with \(F_\theta = -\partial U_\theta / \partial \theta_{ijk}\). The explicit expression for \(\nabla_i \cos\theta_{ijk}\) is algebraically long, so in practice one reuses known formulas from the literature or trusted implementations.
One must also be careful with units: some force fields use degrees in the input files and convert internally to radians when evaluating the potential.
For this, we then need \( \nabla_i \cos\theta_{ijk}\). Eq. 7 of J. Chem. Phys. 146, 226101 (2017)14 offers a compact form.
Warning
\(F_i\) is singlular if \(\theta_{ijk}=0^\circ\) or \(180^\circ\) if \(F_\theta\) is not zero at these points!
2.4.2.1. Harmonic#
The harmonic angle potential mirrors the harmonic bond:
Here:
\(\theta_0\) is the equilibrium angle (straight line: \(180^\circ\), tetrahedral: \(\approx 109.5^\circ\), etc.),
\(k_\theta\) is the angle force constant, controlling how stiffly the molecule resists bending away from \(\theta_0\)
Harmonic angle potentia fixes average angle around set point of \(\theta_0\). Like with the harmonic bond potential, whatch for the factor of two in the spring constant.
Interpretation.
Near \(\theta_0\) the distribution of angles is approximately Gaussian.
Larger \(k_\theta\) means narrower fluctuations (stiffer angle).
The same “factor of \(1/2\)” remark as for harmonic bonds applies here: check whether your simulation package includes it in the definition of \(k_\theta\) or not.
2.4.2.2. Cosine#
A common alternative that avoids computing \(\arccos\) is a cosine angle potential. One simple form is:
Minimum is when the particles are in colinear \(\theta =180^\circ\) arrangement. There are forms with a set angle different from that, by using \(k (1 - \cos{(\theta - \theta_0)}) \) as functional form.
More generally, we can allow an arbitrary equilibrium angle by shifting:
Here the minimum is located at \(\theta = \theta_0\). In both cases the potential depends smoothly on \(\cos\theta\), which is straightforward to compute from the dot product of bond vectors.
Practical point. Cosine angle forms are often preferred in coarse-grained models because the force expressions are simpler and cheaper than those obtained from a harmonic potential written in terms of \(\theta\) itself.
2.4.2.3. Cosine squared#
A third popular choice is the cosine-squared potential:
This form has several nice properties:
It has a minimum at \(\theta = \theta_0\).
The potential is symmetric around \(\theta_0\) when expressed in terms of \(\cos\theta\).
In some cases it naturally produces two symmetric minima (e.g.\ near \(\theta_0\) and \(\pi-\theta_0\)) when combined with other constraints.
Cosine-squared forms appear in various coarse-grained force fields where the angle potential is expressed entirely in terms of \(\cos\theta\).
Watch out for the exact definition of the parameters, sometimes \(1/2\) is included in the definition of \(k\).
Note
For specific models and force fields, always check if the angles are given in degree or radians.
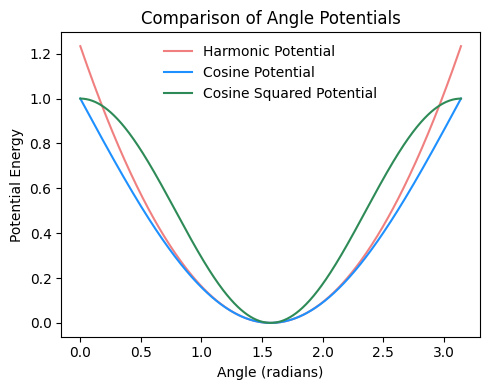
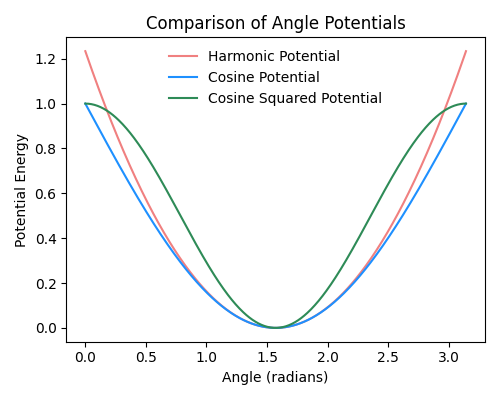
Fig. 2.18 Comparison of harmonic, cosine, and cosine-squared angle potentials, all with an angle constant \(k=1\) and a set angle \(\theta_0\) of \(\pi/2\).#
2.4.3. Dihedral Potentials#
Torsional or dihedral angles are defined as shown in Fig. 2.19. Dihedral (torsional) angles involve four atoms \(i,j,k,\ell\). They are defined via the angle between the two planes spanned by \((i,j,k)\) and \((j,k,\ell)\) (see Fig.~Fig. 2.19). Equivalently, one can ``look down’’ the central bond \(j\)–\(k\) and measure the rotation of atom \(i\) relative to atom \(\ell\).
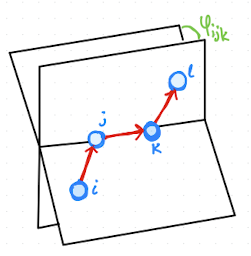
Fig. 2.19 Definition of dihedral or torsional angle.#
Angle between two plane (A,B) normals:
\( \vec{n}_A = \vec{r}_{ij} \times \vec{r}_{jk} \)
\( \vec{n}_B = \vec{r}_{jk} \times \vec{r}_{kl} \)
Then the angle is
\(\varphi_{ijkl} = \frac{\vec n_A \cdot \vec n_B}{|n_A||n_B|}\)
Similar to the angle, alternative definitions exist, \(\psi = \pi -\varphi\).
To compute dihedral forces on four particles, we need to compute
with: \(F_\phi = -\partial U_\phi / \partial \phi_{ijkl}\).
The explicit expressions for the gradients are rather involved and are usually taken from standard references rather than derived from scratch each time.
Singular configurations. Because of the \(1 / \sin\phi_{ijkl}\) factor, dihedral forces can become singular at \(\phi_{ijkl}=0^\circ\) or \(180^\circ\) if \(F_\phi\) is nonzero there. Similarly, if either of the adjacent bond angles \(\theta_{ijk}\) or \(\theta_{jk\ell}\) becomes \(0^\circ\) or \(180^\circ\), the two planes become ill-defined and the dihedral angle is degenerate. Realistic force fields and simulation conditions are chosen to avoid such problematic configurations or to make the potential smooth as these limits are approached.
This results in some involved computations, see Allen & Tildesley2 for a step by step derivation in Appendix C.
Warning
The same singularities as for the angle forces show up at \(\phi_{ijkl}=0^\circ\) and \(180^\circ\) but one can eliminate them (details in Bondel, Karplus, J. Comput. Chem., 17: 1132-1141, 1996)4.
However, \(\phi\) becomes also degenerate if \(\theta_{ijk}\) or \(\theta_{jkl}\) approaches \(0^\circ\) or \(180^\circ\) because the two planes to define the dihedral cannot be defined in these cases. This is important for models with “soft” interactions, where these configurations are possible, e.g. coarse-grained models.
2.4.3.1. Periodic#
A very common torsional form is the simple periodic function:
Used in the CHARMM force field
where:
\(k_\phi\) controls the barrier height,
\(n\) is the multiplicity (number of minima per \(360^\circ\)),
\(\delta\) is a phase shift that moves the minima to the desired dihedral angles.
This form is used, for example, in the CHARMM force field.
Example. For a simple alkane C–C–C–C dihedral, one often chooses \(n=3\) to generate three minima (one \emph{trans} and two \emph{gauche}) over \(360^\circ\). The phase shift \(\delta\) is then chosen so that the global minimum corresponds to the chemically preferred conformation.
2.4.3.2. Ryckaert-Bellemans#
The Ryckaert–Bellemans (RB) form is widely used for alkanes and other chain molecules:
The coefficients \(C_n\) are parameters fitted so that the torsion potential reproduces quantum chemistry data or experimental observables.
Interpretation.
This is effectively a polynomial in \(\cos\psi\) and can approximate fairly complicated periodic shapes.
Shifting by \(\pi\) (using \(\psi\)) is convenient for alkanes where the \emph{trans} conformation near \(180^\circ\) is often of special interest.
The RB form is mathematically closely related to a truncated Fourier series, as we will see next.
2.4.3.3. Fourier#
Used in the OPLS force field. Each term with index \(n\) contributes a periodic component with \(n\) minima or maxima per \(360^\circ\), and the \((-1)^n\) factor flips the sign of the contribution depending on \(n\).
There is a direct mapping between the Fourier coefficients \(C_n\) and the Ryckaert–Bellemans coefficients via trigonometric identities. In practice, force fields may be parameterized in one representation and converted to the other depending on the software.
2.4.4. Improper Dihedrals#
In addition to the dihedrals defined between four bounded particles in a row, there are also so called improper dihedrals, that can be applied to keep planar groups planar, or prevent flipping into mirror images. These are commonly applied between four points in configurations as sketched in Fig. 2.20.
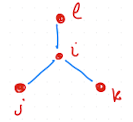
Fig. 2.20 Configuration to which an improper dihedral can be applied to.#
The functional form is often a simple harmonic potential in the improper dihedral angle \(\xi\):
where \(\xi_0\) is usually \(0^\circ\) (to enforce planarity) or some other target angle.
Typical uses.
Keeping aromatic rings or peptide planes planar.
Preventing a stereocenter from flipping into its mirror image.
Stabilizing certain local geometries that are underdetermined by normal bonds and angles alone.
Mathematically, the computation of improper dihedral forces is very similar to that for proper dihedrals; only the connectivity pattern of the four atoms differs.
2.4.5. References#
2.4.6. References#
Allen, Michael P., and Dominic J. Tildesley. Computer Simulation of Liquids. Clarendon Press, Oxford (1987). Appendix C.2: “Calculation of forces and torques.” 2
Howard, Michael P., Antonia Statt, and Athanassios Z. Panagiotopoulos. “Note: Smooth torsional potentials for degenerate dihedral angles.” The Journal of Chemical Physics 146, 226101 (2017). 14
Blondel, Arnaud, and Martin Karplus. “New formulation for derivatives of torsion angles and improper torsion angles in molecular mechanics: Elimination of singularities.” Journal of Computational Chemistry 17, 1132–1141 (1996). 4
Weeks, John D., David Chandler, and Hans C. Andersen. “Role of repulsive forces in determining the equilibrium structure of simple liquids.” The Journal of Chemical Physics 54, 5237–5247 (1971). 45
Kremer, Kurt, and Gary S. Grest. “Dynamics of entangled linear polymer melts: A molecular-dynamics simulation.” The Journal of Chemical Physics 92, 5057–5086 (1990). 17
GROMACS development team. GROMACS Reference Manual: “Bonded interactions” section. (Accessed 2025.) 50